
Google's C2S-Scale, Pfizer's FLOWR, Virtual Lung Screening Trial 🚀
Health Intelligence (HINT)

2025-04-21
🚀 (I am a Houston Rockets fan)
New Developments in Research
C2S-Scale: Scaling LLMs for Universal Single-Cell Analysis
Researchers from Yale and Google introduced C2S-Scale, a family of large language models tailored to single-cell RNA sequencing (scRNA-seq) data by representing cellular expression as natural language.
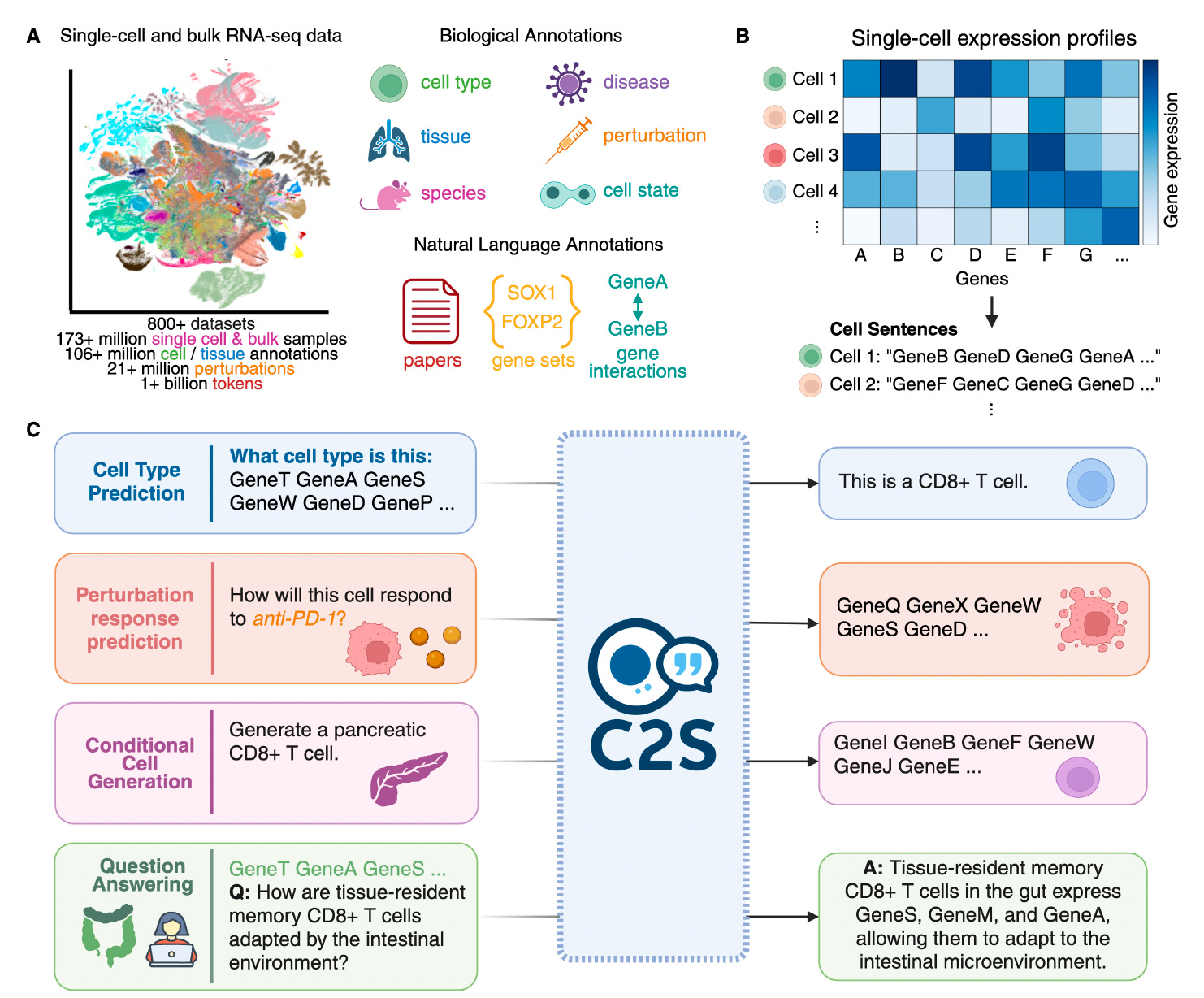
Building on the Cell2Sentence framework, the team trained models up to 27 billion parameters using over a billion tokens from gene expression, biological text, and metadata. This approach allows C2S-Scale to outperform both specialized single-cell models and general-purpose LLMs across a broad spectrum of biological tasks.
Converted gene expression data into ranked “cell sentences” of gene names, enabling language models to process transcriptomic profiles without modifying model architecture or tokenizers.
Trained on a massive corpus of 50 million human and mouse cells paired with annotations and paper abstracts, allowing for rich multimodal learning and downstream generalization across cell types, tissues, and experimental conditions.
Achieved state-of-the-art results on single-cell tasks such as cell type annotation, perturbation response prediction, spatial niche inference, and dataset-level interpretation, outperforming scGPT, GPT-4o, and others.
Applied reinforcement learning via Group Relative Policy Optimization (GRPO) to fine-tune the model on question answering and perturbation tasks, significantly improving alignment with biologically accurate responses and boosting generalization to unseen settings.
C2S-Scale sets a new standard for LLMs in biology, uniting natural language and transcriptomic data to drive scalable, interpretable, and biologically informed single-cell analysis.
MedReason: Enhancing Medical Reasoning in LLMs with Knowledge Graphs
A new study introduces MedReason, a dataset and fine-tuning method that improves factual step-by-step medical reasoning in large language models (LLMs). By grounding reasoning chains in a structured knowledge graph, MedReason generates clinically valid explanations for medical QA tasks.
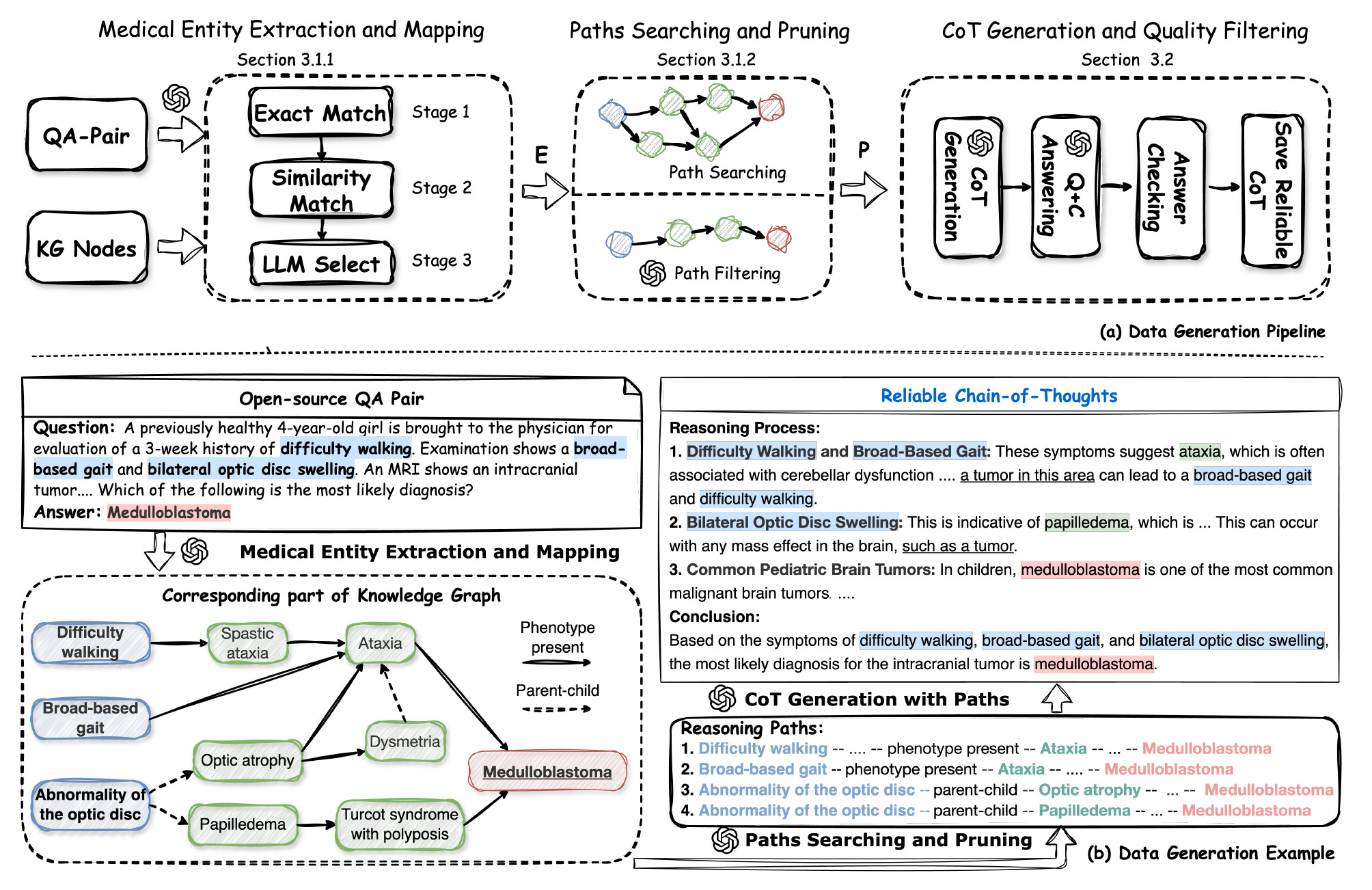
The authors also present MedReason-8B, a fine-tuned LLM that sets new benchmarks on multiple medical datasets.
Generated over 32,000 high-quality question-answer pairs with detailed reasoning steps by mapping medical entities to PrimeKG and extracting verified reasoning paths between them.
Incorporated a quality filtering stage that discards reasoning traces that do not lead to correct answers, improving average benchmark performance by 1.1%.
Improved accuracy across eight medical QA datasets when used to fine-tune base models like LLaMA3 and Mistral, with performance gains of up to 13.7% over baseline models.
Outperformed prior state-of-the-art medical reasoning models on challenging benchmarks such as MedBullets, MedXpert, and Humanity’s Last Exam, with experts from seven medical specialties preferring MedReason’s explanations in head-to-head comparisons.
MedReason highlights the value of combining structured biomedical knowledge with language models to produce clinically sound, interpretable reasoning in medical AI.
Multimodal Cell Maps as a Foundation for Structural and Functional Genomics
A large team of researchers has built a multimodal map of human cell architecture by integrating biophysical protein interactions and immunofluorescence imaging for over 5,100 proteins.
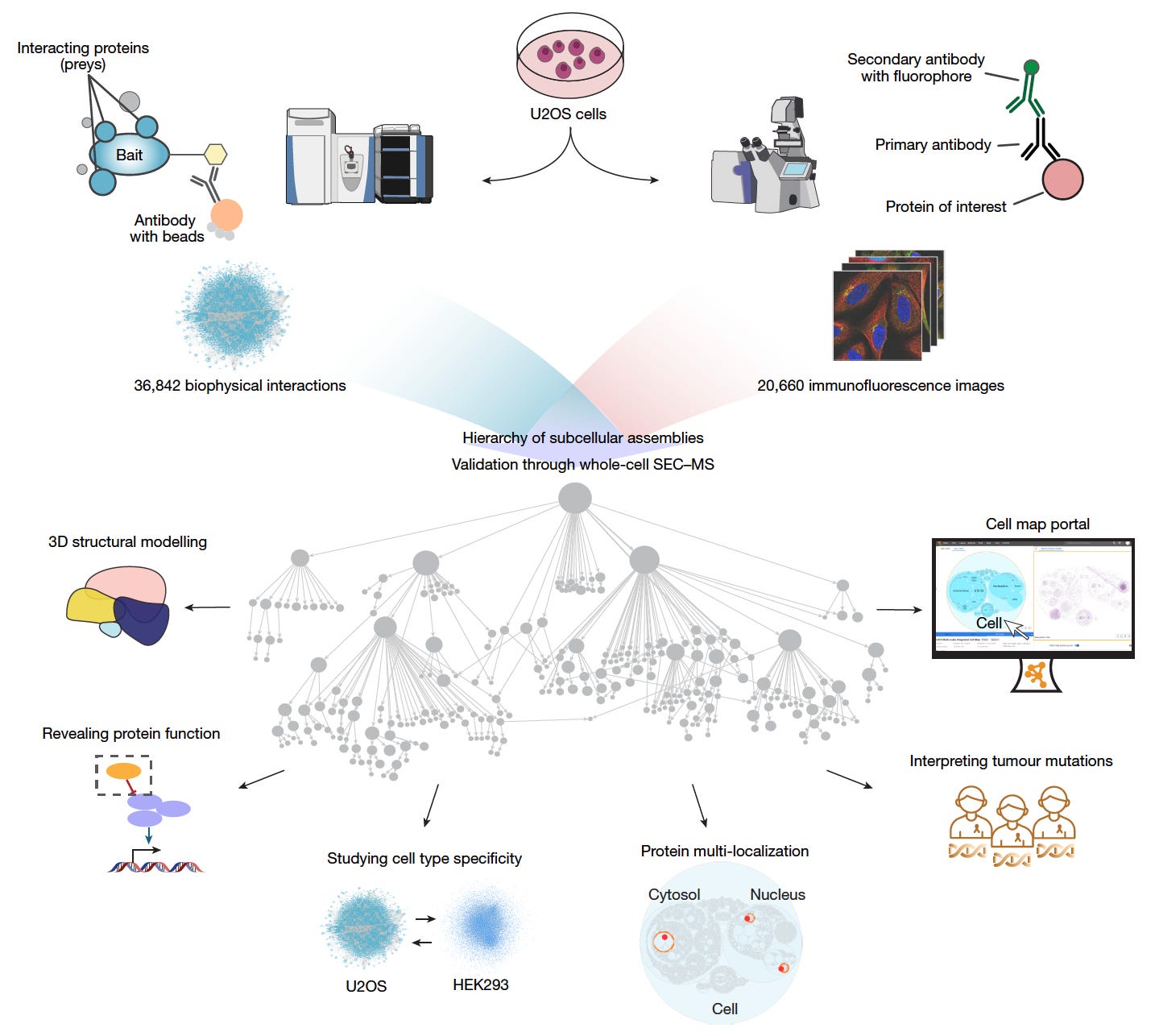
The resulting map resolves 275 molecular assemblies across four orders of magnitude in size, enabling discoveries in protein structure, function, and disease associations.
This resource sets a new reference for exploring subcellular organization and its links to cancer biology, protein function, and spatial proteomics.
Integrated affinity-purification mass spectrometry and high-resolution confocal microscopy to create a self-supervised embedding that identified 275 protein assemblies ranging from nanometres to micrometres in size.
Used large language models, including GPT-4, to annotate assemblies with high-confidence functional descriptions, naming 104 previously undocumented assemblies with supporting literature and self-assessed confidence scores.
Validated 89 assemblies using whole-cell size-exclusion chromatography mass spectrometry, and resolved 111 novel high-confidence protein structures with AlphaFold-Multimer, including the expanded Rag–Ragulator complex.
Identified 21 assemblies recurrently mutated in 772 pediatric tumors and proposed functional roles for 975 proteins, such as implicating C18orf21 in the RNase MRP complex and DPP9 in interferon signaling.
This multimodal map advances a new standard for cellular reference atlases, integrating diverse data to reveal the molecular logic of cell organization and disease mechanisms.
Neural Models for Detection and Classification of Brain States and Transitions
Researchers have developed a deep learning pipeline to decode and classify brain states and their transitions using local field potential (LFP) recordings in rats under anesthesia.
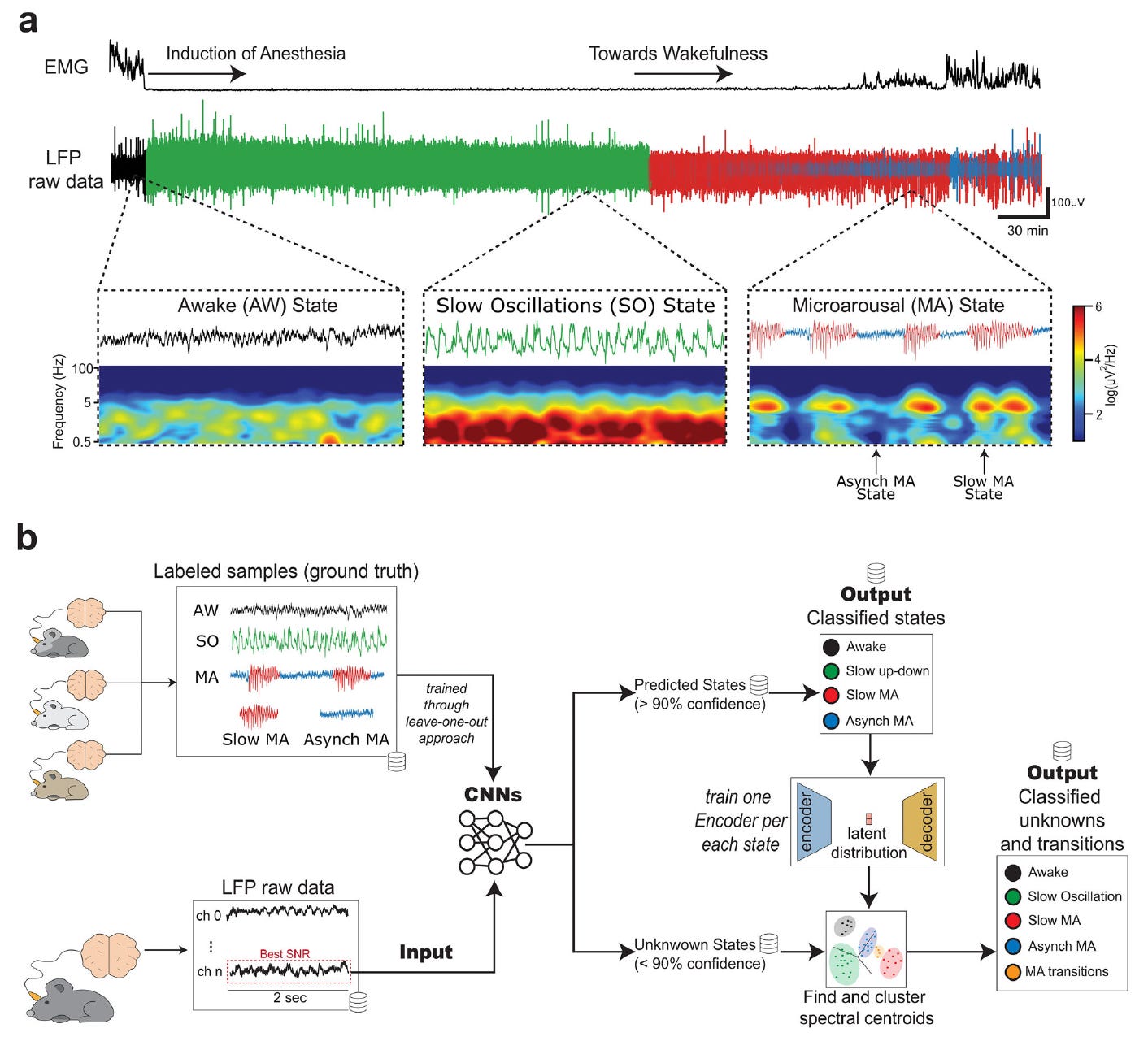
The system combines convolutional neural networks (CNNs) and autoencoder-based clustering to detect transitions between slow oscillations, microarousals, and wakefulness with high confidence. This work provides a valuable tool for basic and clinical neuroscience focused on understanding altered states of consciousness.
Built a dual-CNN model to classify brain states like slow oscillations, microarousals, and wakefulness, achieving up to 96% accuracy for specific states and 91% overall pipeline accuracy.
Integrated an autoencoder-based clustering method to handle low-confidence samples and detect nuanced transitions between microarousal substates, reaching 75% accuracy in transition identification.
Employed a leave-one-out training strategy for generalizability across subjects and sessions, enabling the development of pre-trained models for potential on-device inference.
Demonstrated strong model robustness even on ambiguous data, with confidence thresholds guiding classification and a fallback clustering strategy improving accuracy for subtle state changes.
This model advances brain state decoding by merging invasive electrophysiology with modern neural networks, offering a scalable and interpretable approach for exploring neural dynamics under anesthesia.
Virtual Lung Screening Trial (VLST): A Simulated Clinical Trial for Lung Cancer Detection
In a groundbreaking step toward digitizing clinical research, Duke researchers introduced the Virtual Lung Screening Trial (VLST), an end-to-end in silico study inspired by the National Lung Screening Trial.
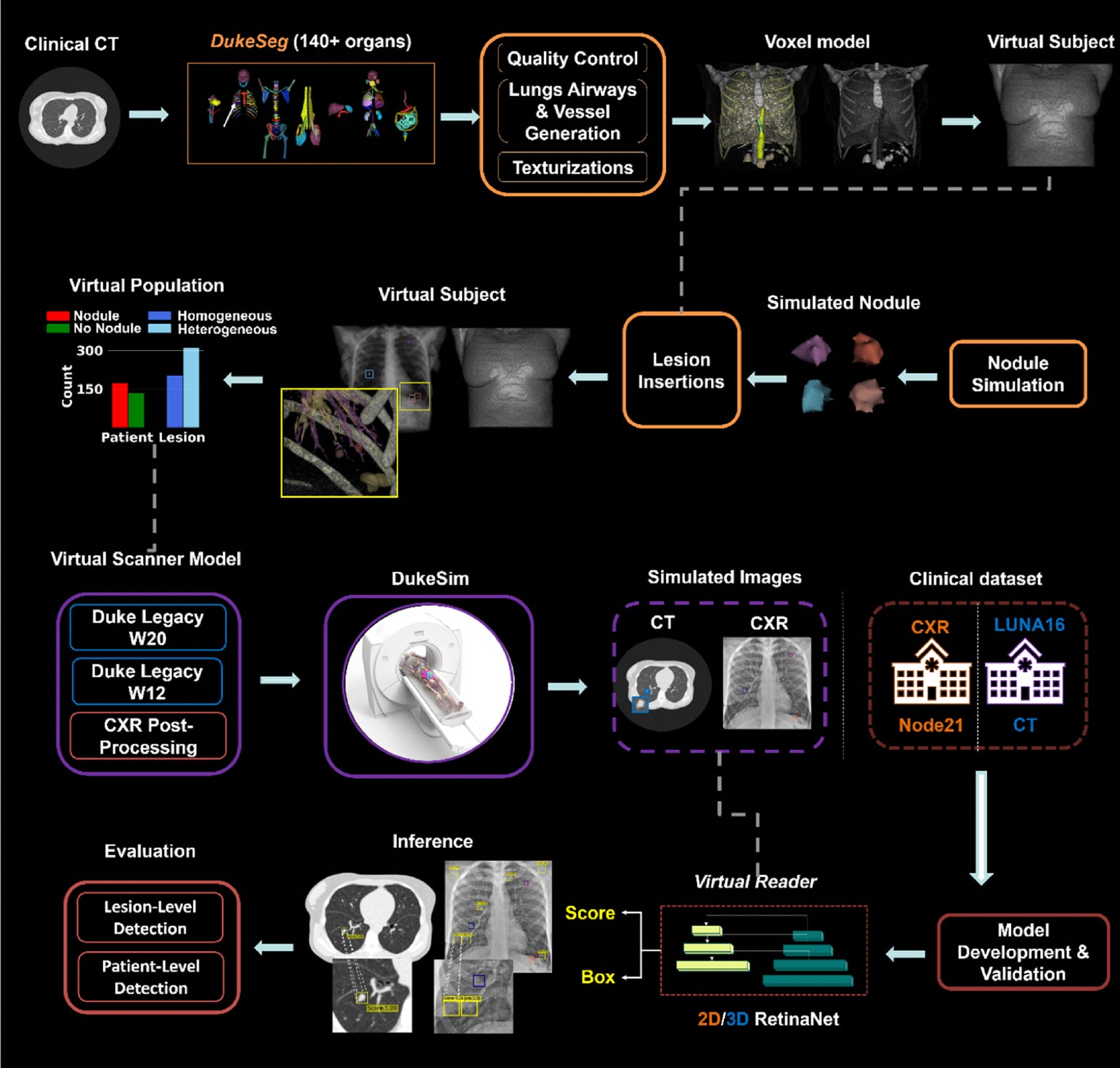
By simulating patients, scanners, and readers, VLST enables safer, faster, and cost-effective evaluations of imaging technologies for lung cancer screening. The study compared CT and chest X-ray (CXR) using AI readers trained on public datasets, achieving diagnostic performance similar to real-world trials.
Created a virtual cohort of 294 patients using anatomically diverse XCAT models, simulating over 500 lung nodules with varying textures to mirror real-world cancer presentations.
Simulated CXR and CT scans using DukeSim, replicating image characteristics from NLST-era scanners, and validated the imaging quality using open-source reconstruction tools.
Deployed AI-based virtual readers (2D/3D RetinaNet models) to detect actionable nodules, achieving an AUC of 0.92 for CT and 0.72 for CXR, closely matching NLST benchmarks.
Demonstrated that CT consistently outperformed CXR across lesion types, with CT AUCs of 0.96 (homogeneous) and 0.89 (heterogeneous), reinforcing the validity of virtual trials as a substitute for early-stage diagnostic evaluation.
VLST marks a pivotal shift in imaging research, establishing a reproducible and ethical framework for simulating clinical trials without exposing patients to real-world risks.
FLOWR: A Fast and Flexible AI Model for 3D Ligand Generation
AstraZeneca and Pfizer researchers introduced FLOWR, a flow matching model for generating 3D ligands conditioned on protein pockets and interactions. Unlike diffusion-based models, FLOWR achieves up to 70× faster inference while improving validity, pose accuracy, and interaction recovery.
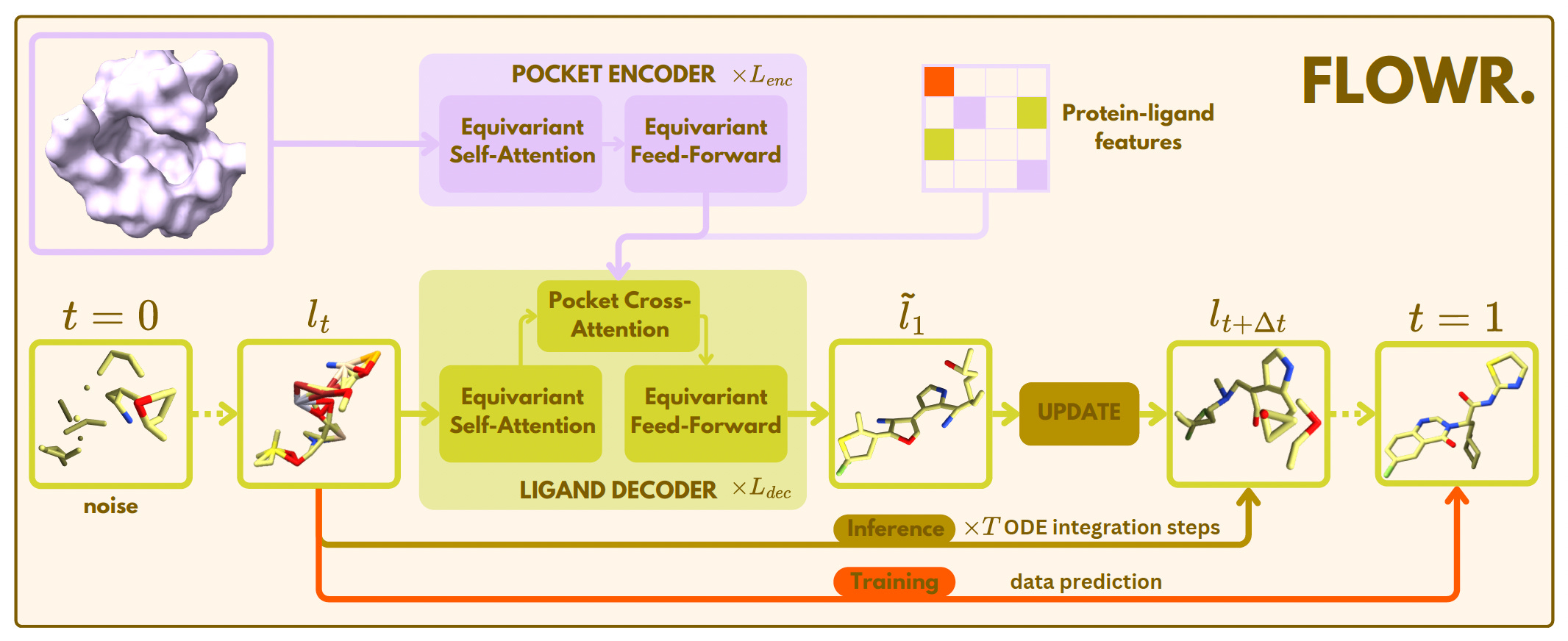
The study also introduces FLOWR.MULTI for fragment-based and interaction-conditional ligand design, alongside SPINDR, a new benchmark dataset with curated, realistic protein-ligand complexes.
Surpassed previous state-of-the-art models (like PILOT) on ligand validity (PoseBusters-validity: 0.92 vs. 0.83), docking score (AutoDock Vina: -6.29 vs. -5.73), and strain energy, while generating chemically realistic ligands up to 70× faster.
Enabled conditional generation with FLOWR.MULTI, supporting tasks like scaffold hopping and interaction inpainting without retraining, achieving a 76.1% interaction recovery rate and improving binding affinity predictions.
Used a single protein pocket encoding to amortize computational cost, drastically reducing inference steps (down to 20) with minimal performance trade-offs.
Developed the SPINDR dataset with 35,666 co-crystal complexes, resolving data quality issues and providing atom-level interaction annotations to improve benchmarking in structure-based drug discovery.
FLOWR and FLOWR.MULTI push the boundaries of generative modeling in drug discovery, offering scalable, accurate, and versatile tools for de novo and fragment-based ligand generation.
Love Health Intelligence (HINT)? Share it with your friends using this link: Health Intelligence.
Want to contact Health Intelligence (HINT)? Contact us today @ lukeyunmedia@gmail.com!
Thanks for reading, by Luke Yun