


EvoWeaver, ProGen3, Meta-Prediction for Coronary Artery Disease Risk 🚀
Health Intelligence (HINT)

2025-04-28
🚀
EvoWeaver: Large-Scale Prediction of Gene Functional Associations from Coevolutionary Signals
A new study presents EvoWeaver, a machine learning framework that integrates 12 coevolutionary signals to predict functional associations between genes across 8,564 genomes.
Unlike traditional annotation methods that rely heavily on prior knowledge, EvoWeaver leverages genomic sequences alone to infer gene function, helping to address the annotation inequality in protein databases.
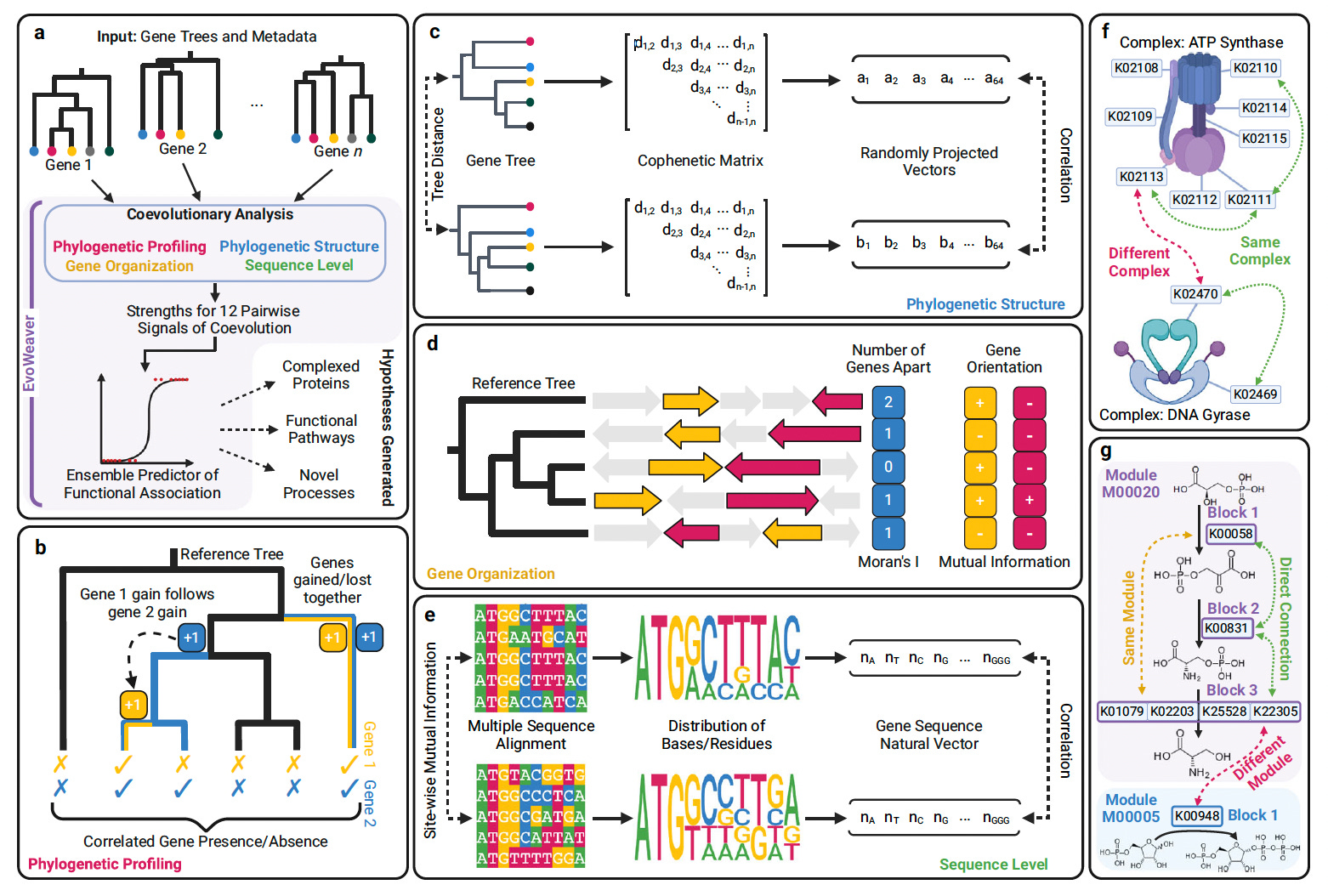
EvoWeaver demonstrated strong performance in reconstructing biochemical pathways and protein complexes, achieving predictive accuracy comparable to the STRING database without relying on external data sources.
Integrated four categories of coevolutionary analysis (phylogenetic profiling, phylogenetic structure, gene organization, and sequence level) using 12 optimized algorithms to enhance scalability and accuracy.
Employed ensemble machine learning models (logistic regression, random forest, neural networks) to combine semi-orthogonal signals, achieving an AUROC of 0.955 for predicting adjacent genes in biochemical pathways.
Outperformed individual coevolutionary methods and rivaled STRING predictions for gene functional associations, even without relying on prior knowledge such as literature mining or curated databases.
Applied EvoWeaver to 1,545 gene groups across 8,564 genomes, identifying missing connections in established databases and generating high-confidence hypotheses for undiscovered functional links, including evidence-supported predictions between genes like B3GNT5 and ST6GAL1 in human glycosylation pathways.
By uniting diverse evolutionary signals, EvoWeaver offers a scalable, annotation-independent approach to illuminate the functional landscape of uncharacterized proteins across the tree of life.
ProGen3: Scaling Sparse Protein Language Models for Enhanced Protein Design
ProGen3 is a family of sparse protein language models (PLMs) trained on a curated dataset of 3.4 billion full-length proteins, the Profluent Protein Atlas v1.
By scaling up to 46 billion parameters and applying optimized data sampling strategies, ProGen3 pushes the limits of protein generation, fitness prediction, and sequence diversity.
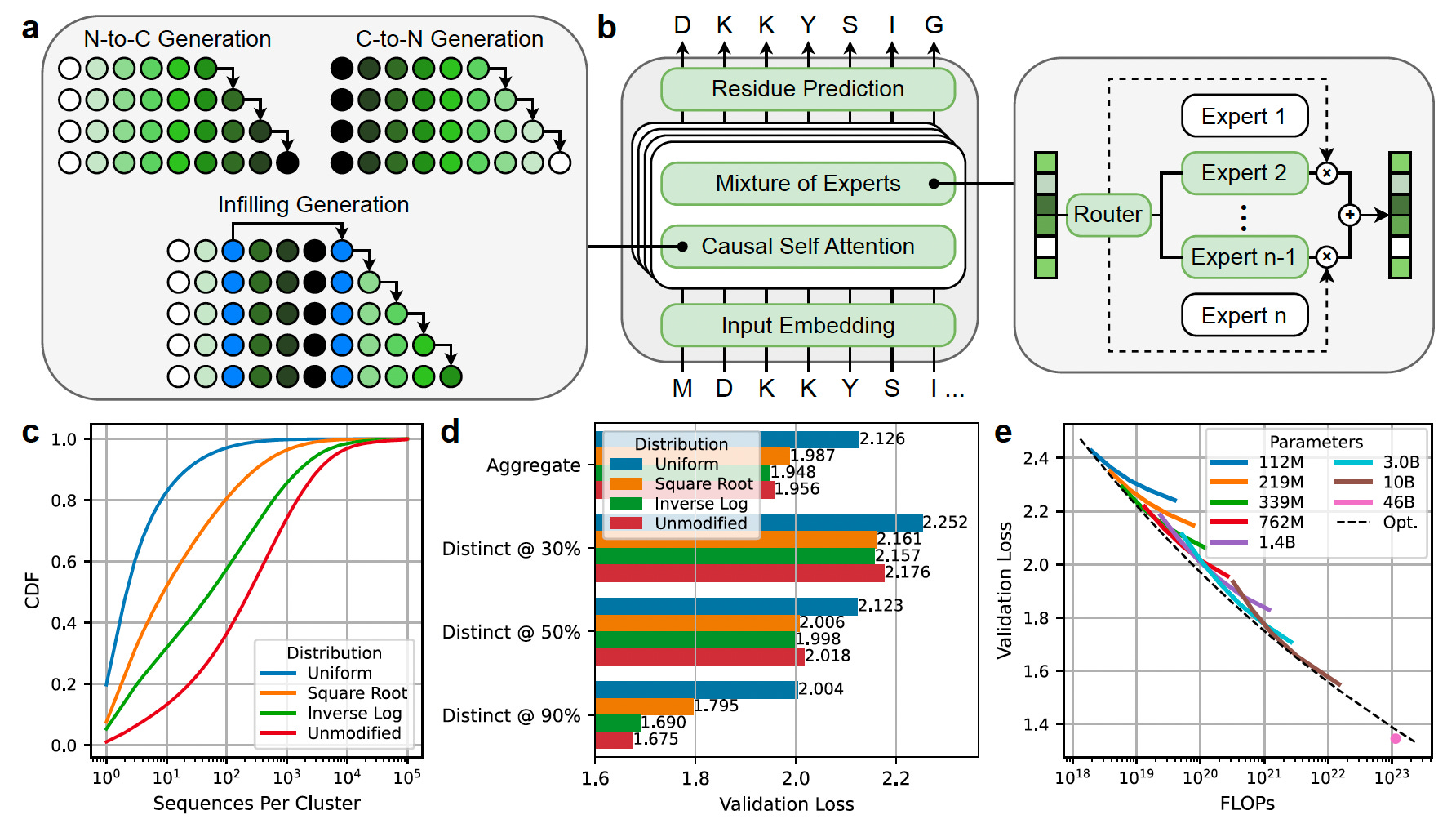
For the first time, researchers experimentally evaluated the impact of model scale on protein viability, demonstrating that larger models generate functional proteins across a broader range of families.
Scaled model parameters from 112 million to 46 billion, leveraging a sparse mixture-of-experts transformer architecture that activates just 27% of parameters per forward pass to improve efficiency without sacrificing performance.
Trained ProGen3-46B on 1.5 trillion amino acid tokens using an "Inverse Log" data sampling distribution, balancing sequence diversity and representation to optimize out-of-distribution generalization.
Demonstrated that larger models generate a wider diversity of valid protein sequences, expressing functional proteins in vitro across more protein families compared to smaller models.
Applied iterative alignment with laboratory data to improve protein fitness prediction and stability generation, with the aligned ProGen3-46B model outperforming or matching state-of-the-art supervised predictors while generating novel, stable protein sequences.
By uniting model scaling, curated datasets, and experimental alignment, ProGen3 establishes a foundation for flexible and scalable protein design across medicine, agriculture, and industry.
scMultiSim: A Versatile Simulator for Single-Cell Multi-Omics and Spatial Data
This is a comprehensive simulator designed to generate synthetic single-cell data across multiple modalities, including gene expression, chromatin accessibility, RNA velocity, and spatial cell locations.
Unlike existing simulators that often focus on one or two biological factors, scMultiSim integrates gene regulatory networks (GRNs), cell–cell interactions (CCIs), chromatin accessibility, and cell identity, providing realistic multi-omics data for benchmarking computational methods.
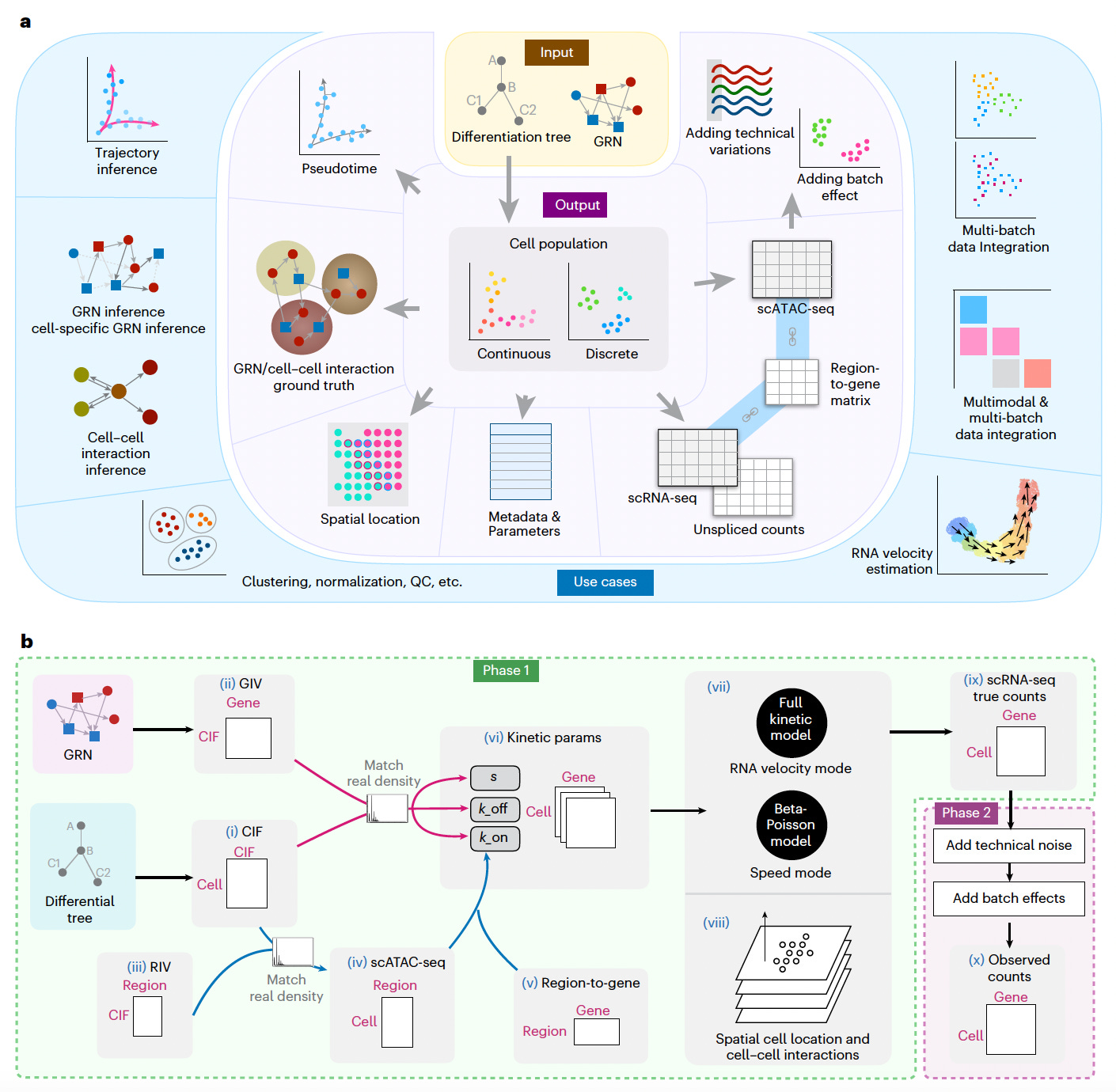
By modeling both biological complexity and technical noise, scMultiSim enables the evaluation of a wide range of single-cell genomics tools, from data integration to GRN and CCI inference.
Simulated multimodal datasets that incorporate biological factors such as GRNs, CCIs, chromatin accessibility, and RNA velocity, while allowing users to control the effect of each factor on the output data.
Validated biological effects in generated datasets, demonstrating realistic gene module correlations guided by GRNs, spatial patterns driven by CCIs, and consistent cross-modality relationships between scRNA-seq and scATAC-seq data.
Benchmarked a broad array of computational methods, including mosaic data integration (e.g., Seurat-bridge, UINMF, Cobolt), GRN inference (e.g., PIDC, GENIE3, CellOracle), and CCI inference (e.g., SpaOTsc, COMMOT), highlighting scMultiSim’s versatility across analytical tasks.
Matched the statistical properties of real single-cell datasets (e.g., 10x Genomics Multiome PBMCs, MERFISH), achieving comparable distributions in key metrics such as library size, zero counts, and gene expression variability.
scMultiSim provides a flexible, biologically informed framework for generating multi-omics and spatial single-cell data, enabling robust benchmarking and development of computational tools across the expanding landscape of single-cell genomics.
CHIMERYS: A Unified Approach for Deconvoluting Chimeric Proteomics Data
CHIMERYS is a novel spectrum-centric algorithm that unifies the analysis of proteomics data across data-dependent (DDA), data-independent (DIA), and parallel reaction monitoring (PRM) methods.
By applying non-negative regularized regression and leveraging deep-learning-based fragment ion predictions, CHIMERYS improves the identification and quantification of peptides from complex, chimeric spectra.
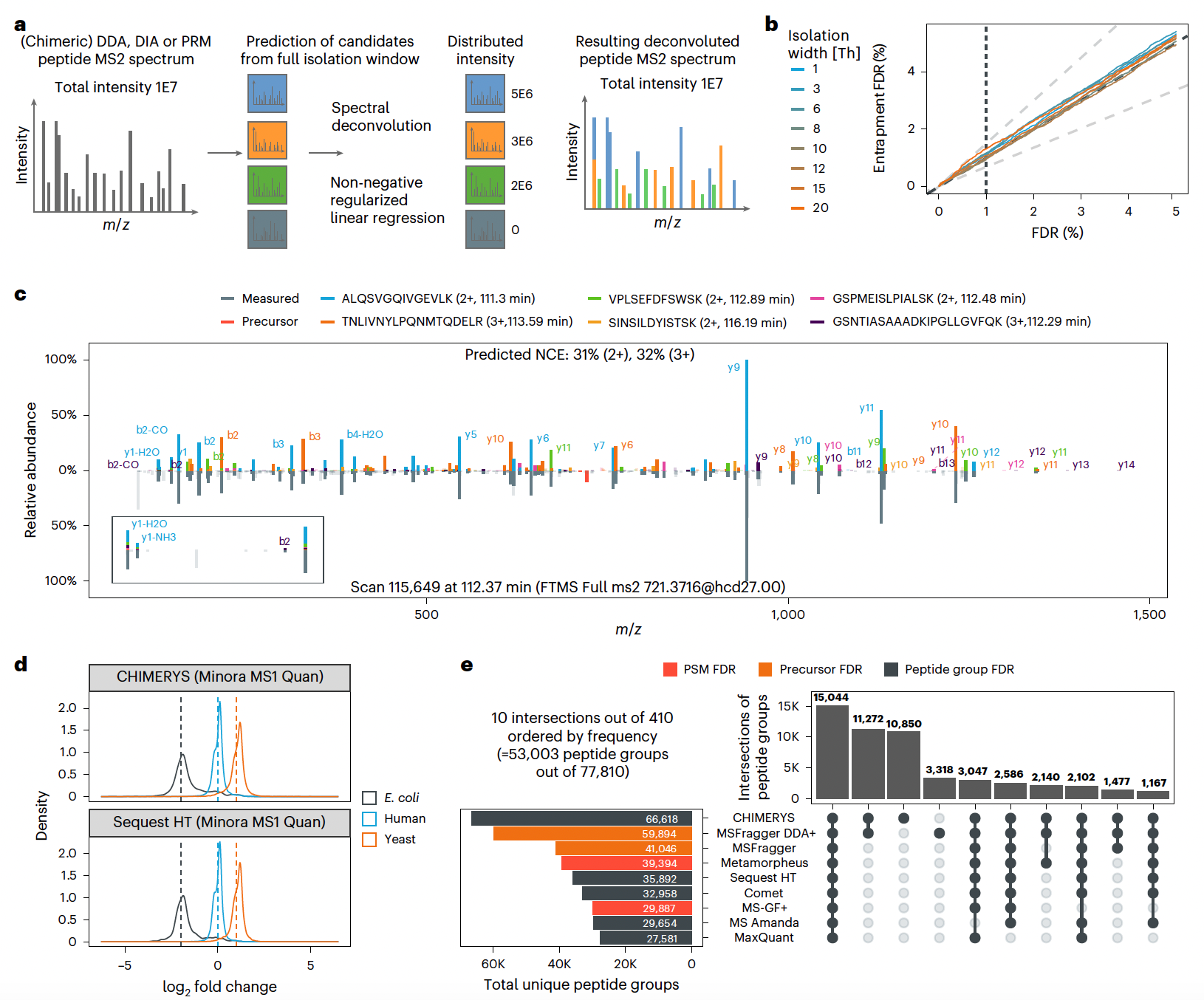
CHIMERYS significantly enhances peptide detection sensitivity while maintaining strict false discovery rate (FDR) control, streamlining proteomics workflows across acquisition strategies.
Applied L1-regularized regression to deconvolute MS2 spectra, accurately assigning fragment ion intensities to multiple co-isolated peptides in chimeric spectra across DDA, DIA, and PRM experiments.
Demonstrated higher peptide and protein identification rates compared to eight other search engines, identifying additional low-abundance peptides in HeLa cell digests while maintaining FDR control.
Improved quantification precision and accuracy, achieving a Pearson correlation of 0.99 against Skyline’s gold-standard PRM quantification, and enabled automated processing without manual intervention.
Outperformed DIA-NN and Spectronaut in DIA data completeness and FDR estimation, maintaining robust quantification even in high-complexity datasets, and unlocked deeper biological insights from direct infusion proteomics data (DI-SPA) that other tools could not access.
CHIMERYS provides a scalable, acquisition-agnostic solution for proteomics data analysis, bridging methodological divides and enhancing both sensitivity and quantification fidelity.
AutoCAR: Sparse 3D Cardiovascular Reconstruction from X-ray Angiography for Instantaneous Diagnosis
Researchers introduce AutoCAR, an automated framework for reconstructing dynamic 3D cardiovascular structures from sparse-view X-ray angiography (XA) images.
Unlike conventional methods that rely on manual annotations or dense scanning, AutoCAR integrates pose domain adaptation, sparse backwards projection, and graph optimization to enable accurate vessel modeling under real-world clinical conditions.
This approach allows for efficient diagnosis and guidance during cardiovascular interventions, overcoming the challenges posed by cardiac motion and limited imaging angles.
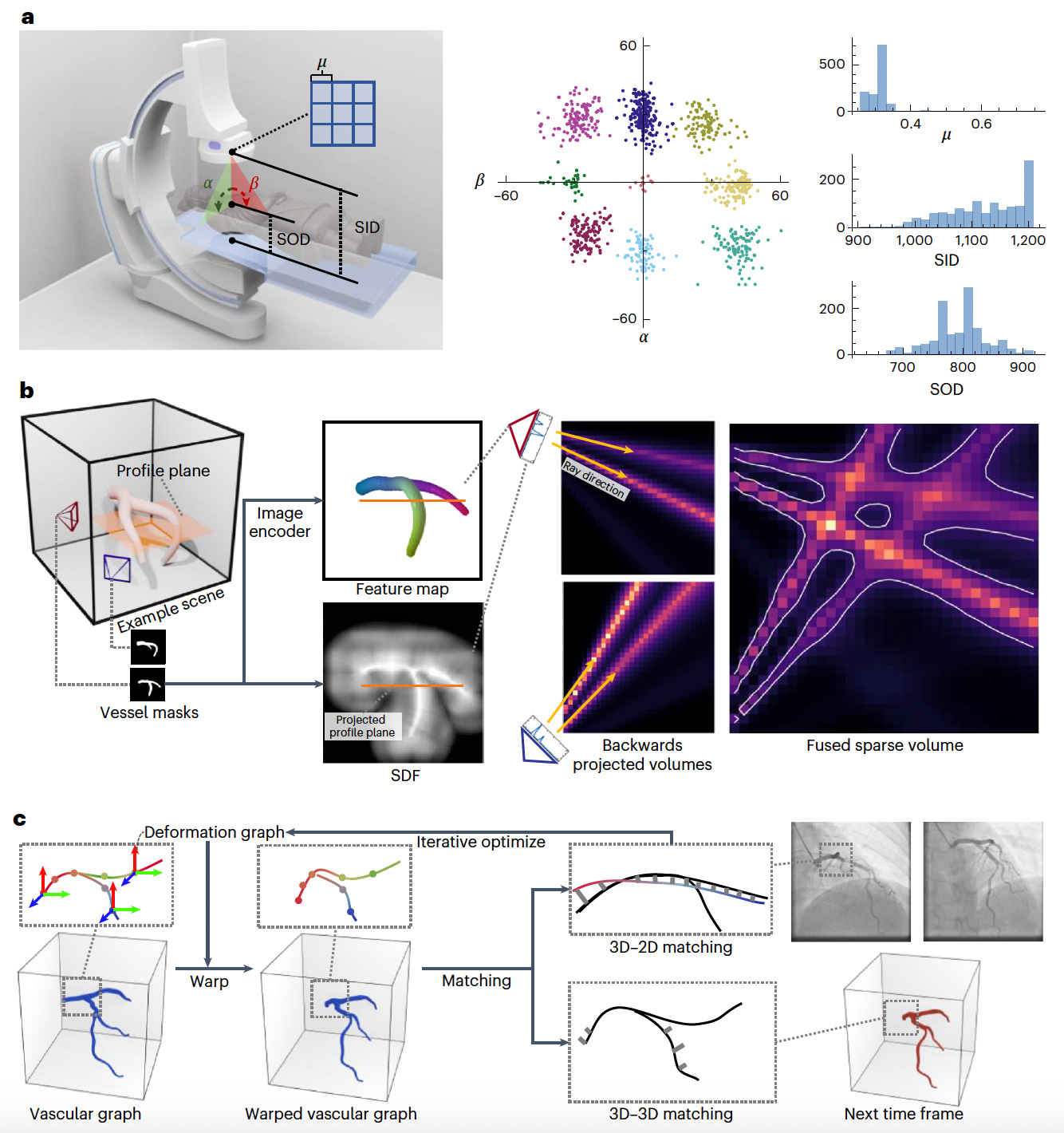
AutoCAR achieves superior performance compared to existing techniques in both synthetic and real-world datasets, offering a robust and transferable solution for dynamic vascular reconstruction.
Incorporated pose domain adaptation to mimic real-world imaging practices by training on synthetic data generated from over 1,200 clinical pose distributions, ensuring robust generalization across varied XA acquisition settings.
Applied sparse backwards projection to exploit the 1D manifold nature of vascular structures, enabling high-resolution 3D reconstruction while minimizing memory and computation demands, outperforming dense projection strategies.
Integrated graph optimization to model the curve-network characteristics of vessels, enhancing temporal coherence and topological accuracy across cardiac cycles during reconstruction.
Achieved 92% correspondence coverage, 1.8 mm end-point error (EPE), and 83% accuracy in real-world XA datasets, surpassing conventional visual hull and learning-based methods while maintaining two orders of magnitude faster processing than human interpretation.
AutoCAR delivers an efficient, real-time solution for 3D cardiovascular imaging, supporting clinical decision-making and paving the way for autonomous navigation in cardiac interventions.
Meta-prediction: Integrating Genetics and Clinical Data for Coronary Artery Disease Risk
Scripps Research introduces a meta-prediction framework that combines genetic and clinical risk factors to forecast 10-year coronary artery disease (CAD) risk.
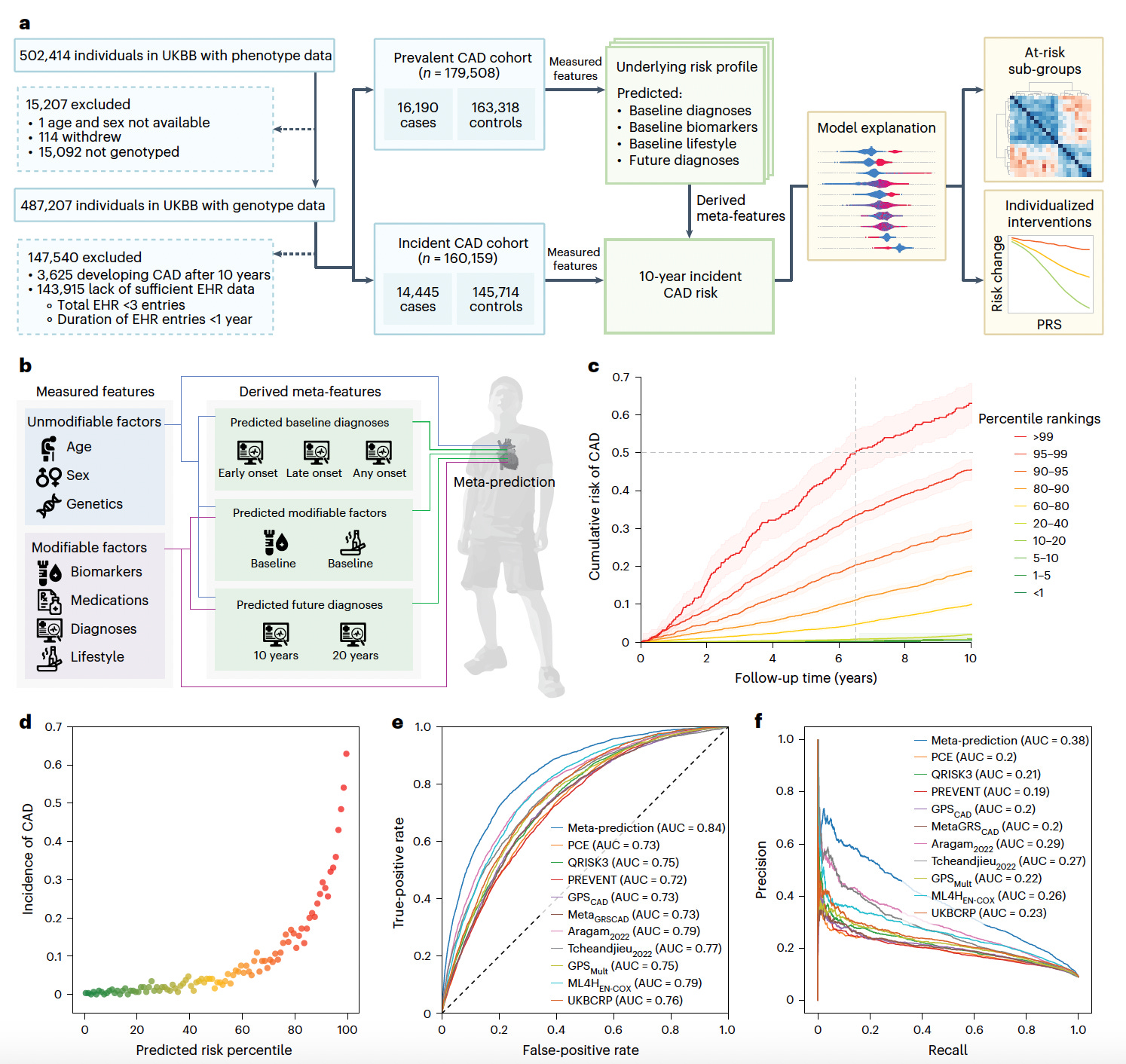
By integrating polygenic risk scores (PRSs) with demographic, clinical, and biometric data, this model achieves personalized and actionable risk assessments. Trained on the UK Biobank and externally validated on the All of Us cohort, this approach significantly outperforms traditional clinical risk scores.
Generated 296 meta-features from 1,431 measured variables, including predictions of future diagnoses and baseline risk factors, enhancing model complexity and interpretability.
Selected 50 final features via Shapley Additive Explanation (SHAP) analysis, including 22 PRSs, 13 measured clinical factors, and 15 meta-features, which together captured diverse genetic and environmental contributors to CAD.
Achieved an AUROC of 0.84 and an AUPRC of 0.38 in the UK Biobank cohort, surpassing conventional clinical risk scores like PCE and QRISK3 by over 10% in discrimination performance and improving reclassification indices.
Validated externally on the All of Us cohort, maintaining an AUROC of 0.81 across diverse ancestries, and demonstrated that genetic risk profiles modulate the benefits of standard interventions like LDL and HbA1c lowering.
By unifying genetic and modifiable risk factors, this meta-prediction model offers a powerful tool for precision prevention of coronary artery disease, particularly in individuals overlooked by traditional risk stratification methods.
Love Health Intelligence (HINT)? Share it with your friends using this link: Health Intelligence.
Want to contact Health Intelligence (HINT)? Contact us today @ lukeyunmedia@gmail.com!
Thanks for reading, by Luke Yun